原作者:Satija Lab
翻译:陈斯杰
原文地址: https://satijalab.org/seurat/v3.1/integration.html
Seurat3提供了一套整合多个单细胞数据集的新方法,这些方法旨在识别出跨多个数据集的shared cell states。这些方法对于实验设计的要求较低——就算数据集采集自不同的个体,使用不同的实验条件,产生自不同的测序技术,甚至来自不同的物种,Seurat3的整合方法都能处理这些数据。
在进行整合时,Seurat3的整合算法首先寻找数据集之间的anchors(固定锚点)。Anchors衡量了两个数据集之间拥有相同cell states的细胞的相关性(correspondence)。借助这些anchors,我们可以将两个数据集整合在一起,将一个数据集上的信息迁移到另一个数据集上。下文中我们将介绍Seurat提供几种整合算法的案例,其中还包括一些2019年manuscript未提及的新功能:
- Standard Workflow
标准Seurat3整合算法案例。本例整合了几种测序技术产生的人胰岛单细胞数据集,还展示了Seurat3如何通过分类算法将已有的cluster labels标注在新收集的被整合数据集上。 - SCTransform
标准Seurat3整合算法的SCTransform升级版( SCTransform是 SatijaLab的新的Normalization算法)。在本例中,我们不仅用它整合了人胰岛数据集,还用它整合了8种技术产生的PBMC数据集,为HCA提供了一个测序技术的benchmark。
Standard Workflow
数据集预处理
导入样例数据集,在样例数据集的metadata中,tech列记录了该细胞使用的测序技术,celltype列记录了细胞类型的注释。我们将panc8中提供的数据集按照测序技术拆分成一个个独立的Seurat Objects,并把它们放到一个R list当中去,按照技术的名字给list中的Seurat Object命名。
在进一步整合之前,我们按照Seurat分析单细胞数据的惯例,对list里各个Seurat Object中存储的对象进行Normalization。
library(Seurat)
library(SeuratData)
InstallData("panc8")
data("panc8")
pancreas.list <- SplitObject(panc8, split.by = "tech")
pancreas.list <- pancreas.list[c("celseq", "celseq2", "fluidigmc1", "smartseq2")]
for (i in 1:length(pancreas.list)) {
pancreas.list[[i]] <- NormalizeData(pancreas.list[[i]], verbose = FALSE)
pancreas.list[[i]] <- FindVariableFeatures(pancreas.list[[i]], selection.method = "vst",
nfeatures = 2000, verbose = FALSE)
}整合来自3种测序技术的数据集
FindIntegrationAnchors函数接受一个Seurat Object的list作为被整合输入项,返回找到的Anchors。本函数常见可调节的参数为dims,通常设置在1:30到1:50之间。 本例中输入的list里包含用三种测序技术测的数据集,我们需要将这三种技术之间的批次效应给去除掉,保留批次效应以外的biological states。
运行IntegrateData函数返回的Seurat Object里有一个新的叫做“integrated”的新assay,这个新assay里包含整合好的(或者说校正好批次的)表达矩阵。整合前原始的基因表达值并没有被丢弃,仍然存留在叫做“RNA”的assay中。
reference.list <- pancreas.list[c("celseq", "celseq2", "smartseq2")]
pancreas.anchors <- FindIntegrationAnchors(object.list = reference.list, dims = 1:30)
pancreas.integrated <- IntegrateData(anchorset = pancreas.anchors, dims = 1:30)整合后我们可以继续进行ScaleData(行归一化),PCA降维,UMAP可视化等操作
library(ggplot2)
library(cowplot)
# switch to integrated assay. The variable features of this assay are automatically
# set during IntegrateData
DefaultAssay(pancreas.integrated) <- "integrated"
# Run the standard workflow for visualization and clustering
pancreas.integrated <- ScaleData(pancreas.integrated, verbose = FALSE)
pancreas.integrated <- RunPCA(pancreas.integrated, npcs = 30, verbose = FALSE)
pancreas.integrated <- RunUMAP(pancreas.integrated, reduction = "pca", dims = 1:30)
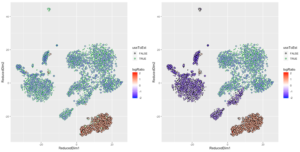
p1 <- DimPlot(pancreas.integrated, reduction = "umap", group.by = "tech")
p2 <- DimPlot(pancreas.integrated, reduction = "umap", group.by = "celltype", label = TRUE,
repel = TRUE) + NoLegend()
plot_grid(p1, p2)
用参考数据集进行细胞类型迁移
除了整合算法,Seurat3还支持迁移算法——将Reference数据集中的数据迁移到Query数据集上。这种迁移和整合有以下几点不同:
– 迁移算法不对Query数据集的基因表达值做校正
– 迁移算法默认把Reference数据集的PCA结构投射到Query的PCA结构上去,而不是用CCA去学一个Query和Reference的共同结构。
在迁移算法中,我们先找Anchors,然后用TransferData函数来对Query数据集中的细胞进行分类。TransferData函数返回一个每个细胞的细胞类别预测结果和预测分数。
pancreas.query <- pancreas.list[["fluidigmc1"]]
pancreas.anchors <- FindTransferAnchors(reference = pancreas.integrated, query = pancreas.query, dims = 1:30)
predictions <- TransferData(anchorset = pancreas.anchors, refdata = pancreas.integrated$celltype, dims = 1:30)
pancreas.query <- AddMetaData(pancreas.query, metadata = predictions)
近期评论