基因表达观测值的分解
SoupX对基因的相对表达的观测值F_{gc}进行背景污染校正。方法作者假设相对表达的观测值可以分解为汤里的游离RNAf_{gs}和细胞里的Genuine表达 f_{gc}两个成分,两个成分按照 \rho_c 和 1-\rho_c 的比例混合。
F_{gc}=\rho_c f_{gs} + (1-\rho_c)f_{gc}
值得注意的是, \rho_c 的下标只有一个 c ,也就是说,对于一个细胞内的不同的基因 g 我们采用同一个污染比例进行校正。我们也可以进一步地弱化这个条件,比如假设所有细胞的背景噪声比例都是一个 \rho (这样并不很合理,nUMI为一万的细胞混一点背景进去,和nUMI为十万的细胞混一点背景进去,背景的比例肯定不一样)。
f_{gs}的估计:
在常规的单细胞数据处理流程中,我们常常设置一个nUMI阈值,把低质量的背景droplets扔掉来提升数据质量。这里我们变废为宝,这些低nUMI的低质细胞,正好可以用来做f_{gs}的估计。文中取了 a <nUMI< b ( a=1,b=10 ) 的droplets,把它们所有的UMI放在一起,用每个基因拥有的UMI所占的比例 \hat{f } _{gs}=n_{gs}/\sum_gn_{gs} 作为其估计量。
\rho_c的估计:
观察 F_{gc}=\rho_c f_{gs} + (1-\rho_c)f_{gc} ,当f_{gc}=0时,即该droplet中某个基因g的UMI全部来自游离RNA时,方程退化为 F_{gc}=\rho_c f_{gs} ,可以利用这个关系估计\rho_c。但是,在细胞级别估计这个值,得到的结果往往是不准确的。
为了让 \rho_c 的估计更准确,我们引入一个假设——对于nUMI相近的细胞,它们的背景污染比例大致是相同的。 这种假设可以在以下实验中得到一个验证:考虑人鼠细胞混合的单细胞实验,这种实验会产生出:1)测后标记为人来源的细胞;2)测后标记为鼠来源的细胞;3)测后标记为doublets的细胞;4)来自人和鼠细胞的游离UMI。可以期待,1)/2)中主要来源的UMI还是人/鼠,少数UMI来自于背景汤。我们可以计数不同的nUMI细胞中的异物种UMI比例来看该假设是否合理。
\rho=\frac{\sum_c\sum_g{n_{gc}}}{\sum_c(N_c\sum_gf_{gs})}
其中N_c是细胞c的nUMI。这内层对g的求和号,并非是对所有的基因求和的,而是每群细胞都有一个不同的基因集合用于求和。这个基因集合是soup-specific的。
选择基因来估计\rho
为了估计背景污染比例,我们需要选择一些“典型”的基因,而不是用所有的基因来估计污染比例\rho。我们选择基因的标准是找表达呈现双峰分布的基因。如果某个基因在各个细胞的表达呈现出双峰,那么其中一个峰代表真实表达此基因的细胞,另一个峰则代表不表达该基因但裹挟了汤中游离UMI的细胞。我们对每个基因计算 F_{gc}/f_{gs} ,其中 F_{gc}是每个基因表达量的观测值, f_{gs} 是某个基因在背景汤中表达量的估计值,可以用前面的公式 \hat{f } _{gs}=n_{gs}/\sum_gn_{gs} 来计算。计算了比值后,我们首先排除掉一些基因——如果满足 F_{gc}/f_{gs}<1的细胞的比例不到10%,那就把这个基因去掉;然后把这些基因按照 log10(F_{gc}/f_{gs}) 大小排序。下图展示的是一些基因的log10(F_{gc}/f_{gs}) 的分布图。
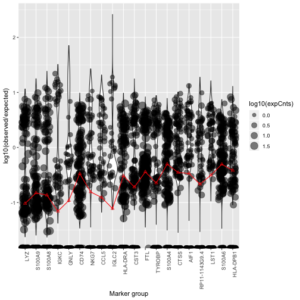
运行教程:1.导入数据并估计背景
以10x数据为例,首先制定10x数据CellRanger运行结果的目录,load10x函数会返回一个SoupChannel类型的对象sc,并自动执行estimateSoup()函数,估计出背景表达f_{gs}的值,保存在sc$soupProfile之中。
library(SoupX)
dataDirs = c("SoupX_pbmc4k_demo/")
sc = load10X(dataDirs, keepDroplets = TRUE)
在得到了SoupChannel对象后,背景汤的基因表达也估计好了。我们可以检查一下某个基因的表达是否比背景汤里的期望水平要高。soupMarkerMap函数可以用于可视化某个基因的UMI count是否比背景汤期望值要高。
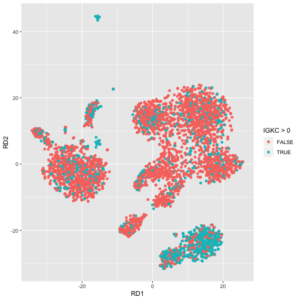
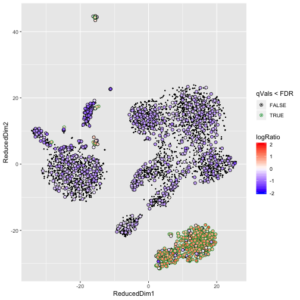
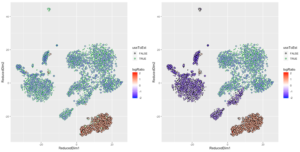
这两幅图中,左图展示的是细胞是否表达IGKC基因。这里面底下一大坨绿色的cluster是B细胞,它们表达IGKC是正常的,但是右边的一大坨其实是T细胞,它们其实是不表达IGKC的,观测到的IGKC更有可能是来自于背景汤的。右图是soupMarkerMap函数展示的IGKC表达的结果。图中的每个散点是一个细胞,黑色的小点表示它们不表达IGKC基因。大一点的点表示该细胞观测到了IGKC的表达,其内部颜色表示 log10(F_{gc}/f_{gs})的值,其边缘的值表示其qVals<FDR(绿色是,黑色否,p-value是假设F_{gc}采样自一个均值为f_{gs}的泊松分布,然后计算观测值的右尾概率得到的,把p-value用BH校正,得到q-value)。我们可以看到,图片下方的B细胞的cluster泛红,表明这一群细胞的IGKC表达值要远高于背景汤的期望值。而右侧的一大坨T细胞中,表达IGKC的那些点,现在的颜色是泛蓝色的,表明它们上面观测到的IGKC表达很可能来自背景汤。
运行教程:2.污染比例估计——基因选择
上一步的背景表达估计没有太大的变数,但是接下来的污染比例估计就对基因选择很敏感。这一步需要人为地指派一些基因,我们将利用这些基因来估计污染的比例。这些基因一般是利用先验生物知识得到的——有一些基因是在一些细胞不表达的,所以可以用来估计污染的比例。比如对于solid tissue来说,红血球是一个普遍而且无法避免的污染,所以血红蛋白基因就是一个非常理想的基因来估计背景污染(血红蛋白基因只在红血球中真实表达)。
因此,我们需要的就是选择丰度较高的、绝对不会在另一些细胞类型表达的基因进行污染比例估计;同时,我们也可以用一些计算手段辅助我们选基因——比如,看看背景汤中高表达的基因。
我们可以这样查看背景汤表达的情况:sc$soupProfile$est中保存的是 f_{gs} 的值, sc$soupProfile$counts中保存的是该基因在所有soup droplet中UMI总计数,排序是按照 f_{gs} 的值从高到低排序的,查看这些基因可能有助于我们选择基因用于估计污染比例。
head(sc$soupProfile[order(sc$soupProfile$est, decreasing = TRUE), ], n = 20)
## est counts
## MALAT1 0.046658138 18345
## TMSB4X 0.015023691 5907
## B2M 0.013790157 5422
## RPS27 0.012431997 4888
## EEF1A1 0.010832216 4259
## MT-CO1 0.010575336 4158
## RPL10 0.010201460 4011
## RPL13 0.010097182 3970
## RPL21 0.009580878 3767
## RPL34 0.009555444 3757
## RPL13A 0.008914515 3505
## MT-CO3 0.008512662 3347
## RPL41 0.008220175 3232
## RPS2 0.008199828 3224
## RPS18 0.008095549 3183
## MT-CO2 0.007579245 2980
## RPL32 0.007220630 2839
## RPLP1 0.007144329 2809
## RPS29 0.007111265 2796
## RPS6 0.007075658 2782
不过从 sc$soupProfile展示的结果来看,背景汤中的很多基因是普遍表达的基因,比如一些核糖体基因和一些线粒体基因。这些基因可能不仅在汤中高表达,在真正的细胞中也高表达,我们不便使用这些基因来做背景污染比例校正。
除了看看汤中高表达的基因,我们还有另一个计算辅助工具plotMarkerDistribution() 。这个函数可以画出来给定的基因集nonExpressedGeneList中基因的log10(F_{gc}/f_{gs})值的分布,如果我们没有显式的传基因集到函数里去,plotMarkerDistribution()会自动调用inferNonExpressedGenes()函数,identify genes that are highly cell specific and so can be used to estimate background contamination。
plotMarkerDistribution(sc)
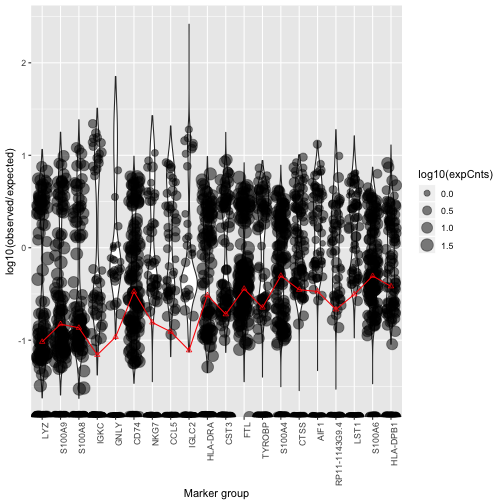
不过值得注意的是,算法自动识别的用于污染比例估计的基因,只是一个备选列表,你不能真的直接用这个列表去做污染比例估计,否则会把污染比例估的偏高。我们可以从这个图中,挑选一些基因用于污染比例估计。
这个图显示了log10(F_{gc}/f_{gs})值的分布,这个值可以用于辅助判断哪些细胞真正地表达了这个基因(图中红线,假设所有细胞都有相同的污染比例)。从图上我们可以看出,有两个免疫球蛋白基因(只有B细胞表达)都在算法推荐的基因之中(IGKC、IGLC2),并且他们的log10(F_{gc}/f_{gs})\approx-1,即F_{gc}\approx10\%f_{gs},也就是说,从这两个基因出发来估计的污染比例是差不多的,都是10%左右。因此,我们选择用免疫球蛋白基因作为估计污染比例的基因。
igGenes = c("IGHA1", "IGHA2", "IGHG1", "IGHG2", "IGHG3", "IGHG4", "IGHD", "IGHE",
"IGHM", "IGLC1", "IGLC2", "IGLC3", "IGLC4", "IGLC5", "IGLC6", "IGLC7", "IGKC")
运行教程:3.污染比例估计——细胞选择
在确认了用于估计背景比例的不表达基因后,我们开始进一步推断,哪些细胞真正表达这些基因,因此不应该用它们来估计背景污染比例,把它们排除在外。我们有两种模式来做这样的事情:无聚类信息和提供聚类信息。
无聚类信息时:
useToEst = estimateNonExpressingCells(sc, nonExpressedGeneList = list(IG = igGenes))
plotMarkerMap(sc, geneSet = igGenes, DR = PBMC_DR, useToEst = useToEst)
## No clusters found or supplied, using every cell as its own cluster.
每一个细胞被看作一个单独的聚类,该函数返回一个矩阵,矩阵指明了哪些细胞(行) 应该使用哪些基因(列)来估计污染比例。下方左图就展示了估计的结果。黑边的点,表示被排除在了背景估计之外,从点的颜色来看,这些点的logRatio比较高,说明免疫球蛋白基因在这群细胞中表达的水平很高,是真实表达的,所以予以排除。SoupX采用偏保守的策略来估计污染比例,它既不从高表达
有聚类信息时:
useToEst = estimateNonExpressingCells(sc, nonExpressedGeneList = list(IG = igGenes),
clusters = setNames(PBMC_DR$Cluster, rownames(PBMC_DR)))
plotMarkerMap(sc, geneSet = igGenes, DR = PBMC_DR, useToEst = useToEst)
在有聚类信息时,如果某个聚类内有一个细胞是真表达那些“”不表达基因”的,这些细胞就不用于污染比例估计了。我们发现,除了下方的一群泛红的B细胞画了黑圈不用作估计,原先左下侧绿圈泛紫的细胞画上了黑圈,它们内部的免疫球蛋白基因表达量,也不用于估计了污染比例了。
运行教程:4.污染比例估计——污染比例计算
所以干了上面那么多事情以后,最重要的一部总算来了,那就是计算污染比例。最后可以看到,也就两个东西最终影响着比例——“不表达基因”和“真表达细胞”。
sc = calculateContaminationFraction(sc, list(IG = igGenes), useToEst = useToEst)
## Estimated global contamination fraction of 4.92%
head(sc$metaData)
## nUMIs rho rhoLow rhoHigh
## AAACCTGAGAAGGCCT 1738 0.04915823 0.04620279 0.05223699
## AAACCTGAGACAGACC 3240 0.04915823 0.04620279 0.05223699
## AAACCTGAGATAGTCA 1683 0.04915823 0.04620279 0.05223699
## AAACCTGAGCGCCTCA 2319 0.04915823 0.04620279 0.05223699
## AAACCTGAGGCATGGT 2983 0.04915823 0.04620279 0.05223699
## AAACCTGCAAGGTTCT 4181 0.04915823 0.04620279 0.05223699
运行教程:5.校正
剩下的内容很简单了,请参看原教程吧。
Reference
SoupX Tutorial
https://cdn.rawgit.com/constantAmateur/SoupX/master/doc/pbmcTutorial.html
SoupX removes ambient RNA contamination from droplet based single cell RNA sequencing data
Matthew D. Young, Sam Behjati https://www.biorxiv.org/content/10.1101/303727v1

近期评论